A new mechanism for reversible proteasome inhibition

In their function as cellular recycling plants, proteasomes fulfill a life-sustaining role in all cells -- including cancer cells. When the proteasomes become inhibited, cells suffocate in their own waste. A reversible proteasome inhibition recently discovered at the Technische Universitaet Muenchen may provide the key to more specific anti-cancer drugs and to controlling rejection reactions in transplantations. The international edition of Angewandte Chemie presents their results online ahead of print, featured as a "hot paper."
What makes cancer cells so dangerous is that they grow in an unregulated way and proliferate much faster than other cells. The proteasome, a large protein complex, plays a key role in this process: By breaking down used proteins for recycling, it clears the way for the next cycle. New hope was spawned several years ago with the discovery that inhibiting proteasomes can be used as a means to put the brakes on cell growth. In the mean time, the first drug using this approach, Bortezomib, generates revenues in excess of one billion U.S. dollars per year. However, it also inhibits other important proteins and thus sets off a host of severe side effects. Hence, there is a worldwide search for alternatives.
A variant of the proteasome, the immuno-proteasome, is a leading actor in another vital process: the immune reaction. The production of human insulin in genetically modified bacteria is a boon for diabetes patients. But calculating required doses and injecting insulin on a daily basis is a considerable burden. The transplantation of intact insulin-producing islet cells from pigs could be a solution, but the immune reaction stands in the way. If physicians were able to curb the immuno-proteasome temporarily, they could possibly get the rejection reaction under control.
In both cases, targeting the intervention as specifically as possible is essential to minimizing the damage caused by side effects. Besides Bortezomib two other proteasome inhibitors – Carfilzomib and Salinosporamide A, derived from a poison produced by a marine bacterium – are already in the human clinical trial stage. In earlier research, Professor Groll's team succeeded in explaining the mechanism of action of both substances. They follow the same principle, which is considerably more specific than Bortezomib in binding at the desired location in the proteasome and thus produce significantly fewer side effects. Yet, when they take effect, they destroy the proteasomes irreversibly. While healthy cells survive by building up a new proteasome, the fast-growing cancer cells suffocate in their own waste and end up in apoptosis.
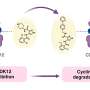
The trick of both of these proteasome blockers is a two-step reaction. The tiny molecule fits a specific site on the proteasome like a key going into a lock. However, the reversible docking at the binding site is followed by an irreversible ring formation that prevents the key from being pulled back out. By choosing another head group, Professor Michael Groll und Dr. Melissa Graewert were able to achieve a reversible ring formation. The new head group contains an aldehyde and a keto group in immediate proximity. They also react with the binding sites on the proteasome by forming a ring. However, the reaction of these two groups is reversible: The key can be removed from the lock and the proteasome can resume its work.
Within the framework of their work supported by the Cluster of Excellence Center for Integrated Protein Science Munich (CIPSM), the TUM scientists succeeded in confirming the assumed mechanism using X-ray structure analysis of inhibited proteasome crystals. It also became clear how this binding mechanism could be developed into a drug with fewer side effects. In addition to the head group, the binding mechanism contains a short chain of amino acids that can be designed to fit into the binding pockets of the proteasome. By varying the amino acids, the binding can be optimized to specifically attack immuno-proteasomes.
"The reversible two-step binding mechanism shown here is unique to proteasomes," says Michael Groll, who holds the Chair of Biochemistry at the Department of Chemistry of the Technische Universitaet München. "This explains the high selectivity and leads us to expect relatively minor side effects. The reversible reaction opens up a much wider field of application. We can now develop these binding mechanisms further in the direction of immunosuppressive agents." It is precisely this challenge the researchers are now taking on, in close collaboration with physicians, through a new SFB-transregional initiative: "Biology of xenogenic cell and organ transplantation – from bench to bedside."
Provided by Technische Universitaet Muenchen